The current JS application is a detector that uses two models, a model "+" that is associated with what we are looking for, and a model "-" that is associated with the background. Both models are represented by a transition matrix that is calculated or trained by using two sequences of observations. Namely, a sequence of observations that is known to belong to a region of interest (model "+") and a sequence of observations that may represent either a random sequence or a sequence other than the sequence "+". Once the model sequences have been used to construct the transition matrices for the two models, they are merged into a single matrix, namely into a log-likelihood matrix (LLM). The log-likelihood matrix represents "the memory", a kind of signature that can be used in some detections. But how? A scanner can use this LLM to search for model-like "+" regions inside a longer sequence called z (the target). To search for such regions of interest, sliding windows are used. The content of a sliding window is examined by verifying each transition with the values from the LLM. Once a transition is associated with a value, it is added to the previous result until all transitions in the content of the sliding window are verified. This results in a main score for each sliding window over z. The positive scores (red) indicate the regions that resemble the "+" model, and the negative scores indicate that the content of the sliding window is different from the "+" model.
Live demo: https://gagniuc.github.io/Markov-Chains-scanner/
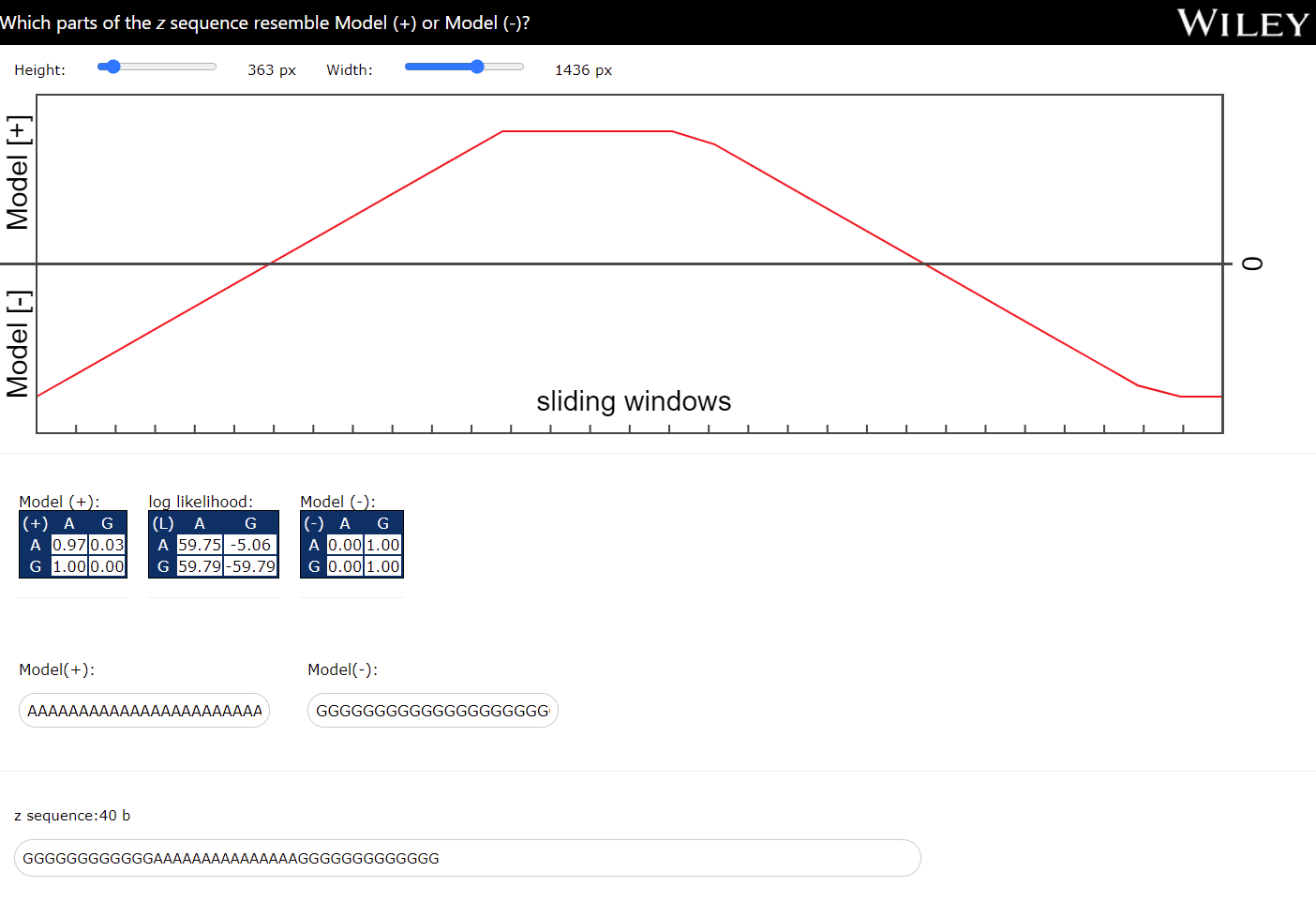
- Paul A. Gagniuc. Algorithms in Bioinformatics: Theory and Implementation. John Wiley & Sons, Hoboken, NJ, USA, 2021, ISBN: 9781119697961.