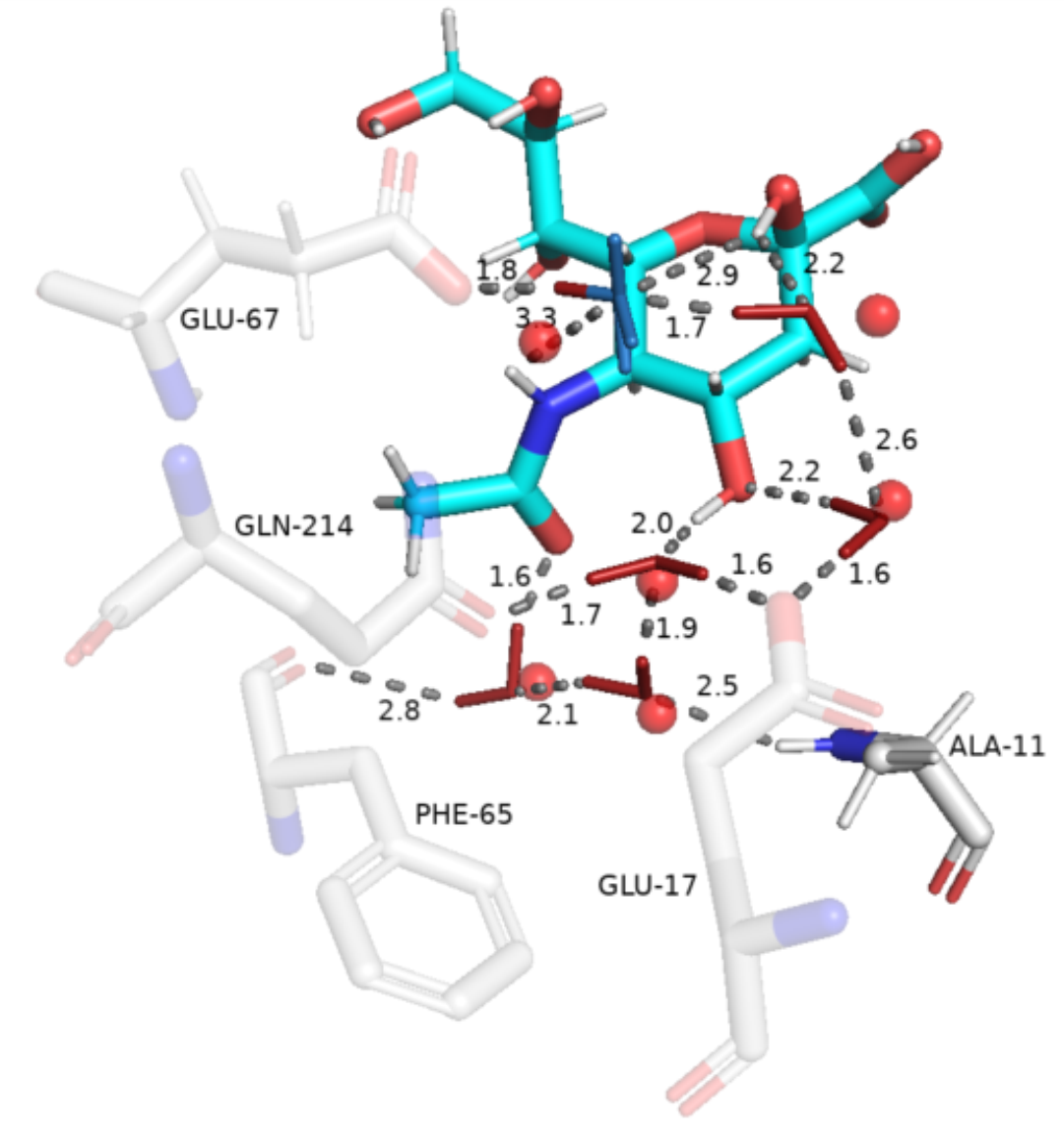
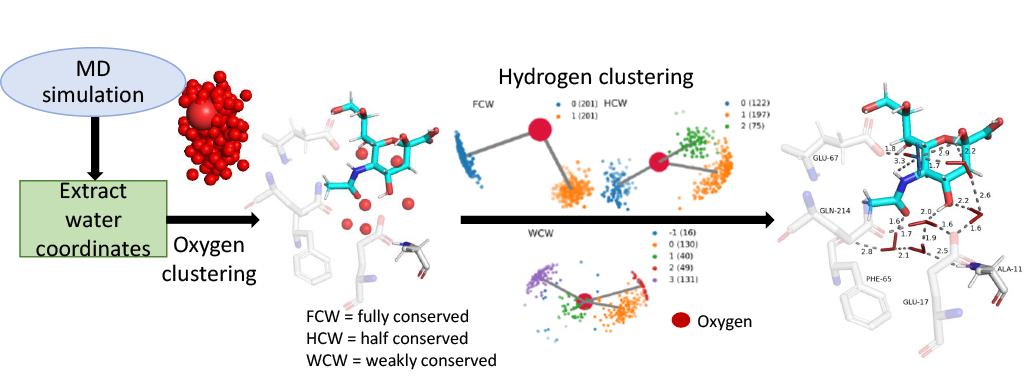
The WaterNetworkAnalysis (WNA) Python package serves as a set of tools for input preparation and further analysis for ConservedWaterSearch (CWS) python package which identifies conserved water molecules from Molecular Dynamics (MD) trajectories.
- Documentation: hosted on Read The Docs
- GitHub repository: source code/contribute code
- Issue tracker: Report issues/ request features
- ConservedWaterSearch: Analysis of conserved waters from simulation trajectories. For docs see here.
For citations and more infromation see ConservedWaterSearch citation.
The easiest ways to install WaterNetworkAnalysis is using conda from conda-forge:
conda install -c conda-forge WaterNetworkAnalysisAlternatively, WNA is also available on PyPi via pip:
pip install WaterNetworkAnalysisPymol is an optional dependency for visualisation and is not present on PyPi, however WNA can be installed and used without it (bar pymol visualisation features). Pymol can be installed using conda:
conda install -c conda-forge pymol-open-sourceFor more information on CWS dependencies also see CWS installation guide.
See documentation for detailed instructions.
AttributeError: 'super' object has no attribute '_ipython_display_'
Some versions of Jupyter notebook are incpompatible with ipython (see here). To resolve install version of ipywidgets<8 using conda:
conda install "ipywidgets <8" -c conda-forgeor pip:
pip install ipywidgets==7.6.0The following example shows how to use WaterNetworkAnalysis to prepare a MD trajectory and analyse the results for determination of conserved water networks.
from WaterNetworkAnalysis import align_trajectory
from WaterNetworkAnalysis import get_center_of_selection
from WaterNetworkAnalysis import get_selection_string_from_resnums
from WaterNetworkAnalysis import extract_waters_from_trajectory
from ConservedWaterSearch.water_clustering import WaterClustering
from ConservedWaterSearch.utils import get_orientations_from_positions
# MD trajectory filename
trajectory="md.xtc"
# topology filename
topology="md.gro"
# aligned trajectory filename
alignedtrj = "aligned_trj.xtc"
# aligned snapshot filename
aligned_snap = "aligned.pdb"
# distance to select water molecules around
distance = 12.0
# align the trajectory and save the alignment reference configuration
align_trajectory(
trajectory=trajectory,
topology=topology,
align_target_file_name=aligned_snap,
output_trj_file=alignedtrj,
)
# define active site by aminoacid residue numbers
active_site_resnums = [111, 112, 113, 122, 133, 138, 139, 142, 143, 157, 166, 167, 169, 170, 203, 231, 232, 238]
# find centre of the active site in aligned trajectory
selection_centre = get_center_of_selection(
get_selection_string_from_resnums(active_site_resnums),
trajectory=alignedtrj,
topology=topology,
)
# extract water coordinates of interest around selection centre
coordO, coordH = extract_waters_from_trajectory(
trajectory=alignedtrj,
topology=topology,
selection_center=selection_centre,
dist=distance
)
# start the clustering procedure
Nsnaps = 200
WC=WaterClustering(nsnaps=Nsnaps)
# perform multi stage reclustering
WC.multi_stage_reclustering(*get_orientations_from_positions(coordO,coordH))
# visualise results with pymol
WC.visualise_pymol(aligned_snap, active_site_ids=active_site_resnums, dist=distance)