
A CLI tool to predict foldability of a peptide sequence. The program accepts a FASTA file with a multiple number of peptide sequences as input.
The script was written in and is intended to be used with Python 3.6; preFold works with 2.7 interpreter but use with caution! You will need a SciPy stack installed to run the program with a full functionality. Windows users must also install colorama package.
Install required packages and a preFold CLI tool (highly recommended together with virtualenv or similar) with:
bash install_cli_tool.sh <python-distribution> # the argument here is the Python interpreter to be used to build preFold
Alternatively, you can install with "pip install ." or load the tool directly from /pprefold/init.py
prefold -i <fasta_file>
This will print out some information about the sequence, disordered regions in CLI and generate .png file showing the predicted foldability of the peptide sequence.
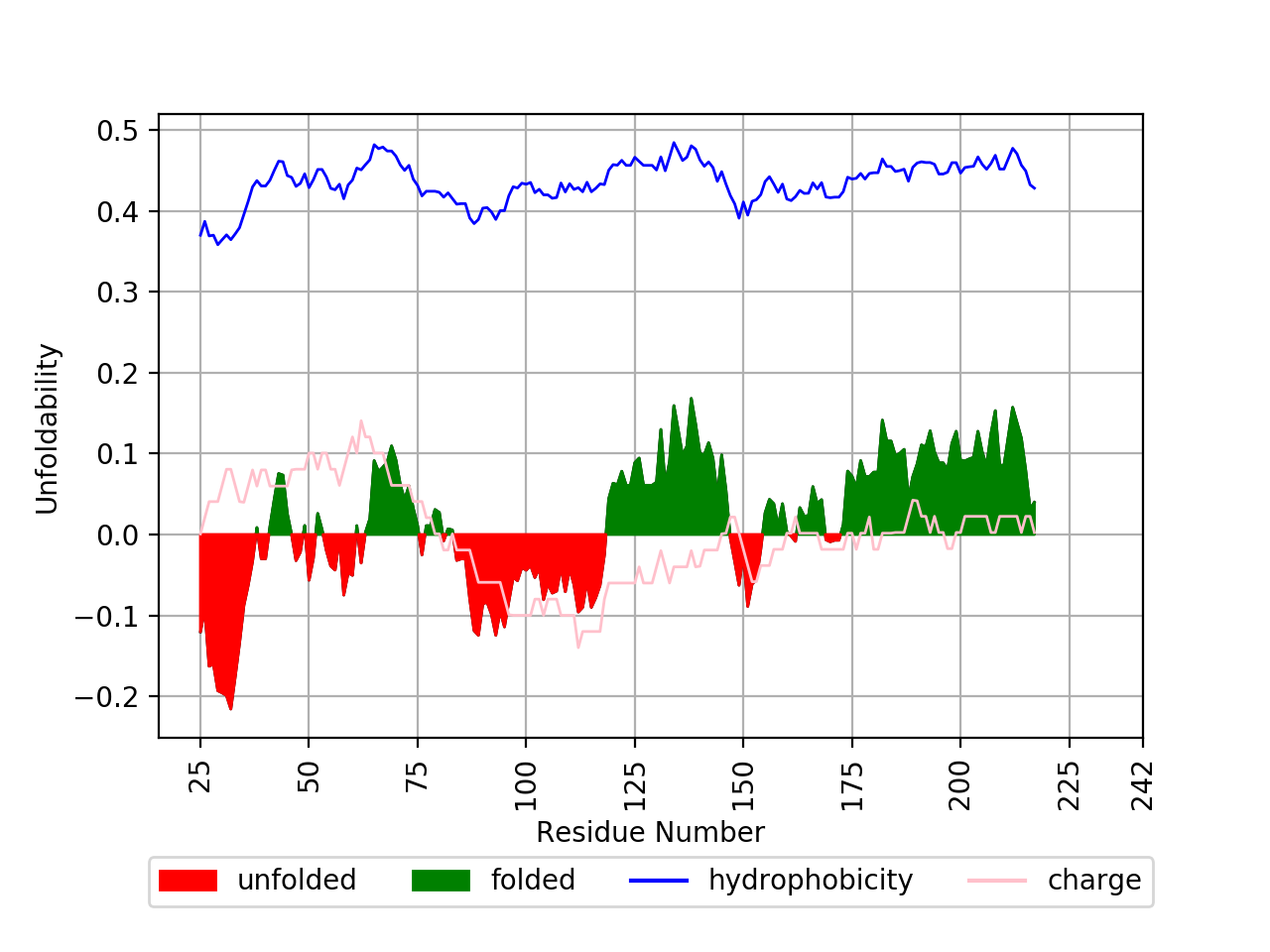
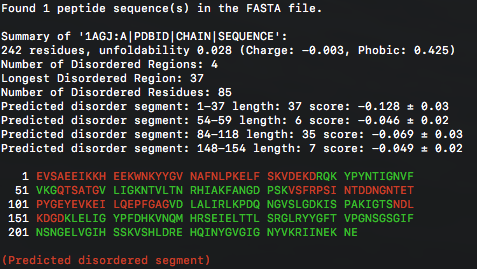
- -i a flag to specify the filename and/or the directory of a file (note that this is the only required argument);
- -ph a flag to specify a pH value to be used in the calculation (default = 7.4);
- -s a flag to specify a step for the sliding window to be used in the calculation (default = 1);
- -k a flag to specify the window size (default = 50);
- -z Specify the flag for charge of the sequence to be plotted on the figure;
- -hb Specify the flag for hydrophobicity of the sequence to be plotted on the figure.
- -ter a flag to exclude N and C terminal charges from the calculation;
- -pka a flag to specify a file containing pKa values of amino acids. Please use the default file as an example for the input file format. Must be located in a datasets folder (default = PKA_DATA_VOET.DAT);
- -b Specify the boundry for calling disordered regions of peptide sequence (default=± 0.005);
- -csv a flag to generate numerical output in .csv format;
- -f Specify the dpi (resolution) of a figure (default = 200).
Please use --help flag for more information on all options and parameters.
This tool is a Python clone of a FoldIndex web app. preFold surpasses the original in accuracy, availability of options and fixed mistakes. Here are listed a few of them:
- Allows the user to modify pH value (this feature is crucial for crystallographers);
- The use of a residue pKa value table of one's preference (default is as provided by Voet & Voet);
- Improved peptide charge plotting (negative values are plotted instead of absolute values as in FoldIndex).
- The boundry line ± 0.005 for calling disordered peptide regions is included in this tool in contrast to the FoldIndex (it seems the FoldIndex authors in the publication mistakenly claimed it to be included). Also, this boundry line can be modified by the user;
- Optional inclusion of N and C terminal charges into the calculation;
- Uses floating point values for increased precision throughout the calculation (i.e the values are only rounded when messages are printed to the user in the CLI);
- Generation of publication quality figures with an option to regulate the resolution of the output figure;
- .pyx file for Cython compilation for performence increase in speed.
- Web app
- The tool does not account for the pertrubed pKa values of residues by the neighbouring residue groups and does not assume any disuphide bridges.
- You might also notice minor differences in results produced by preFold and FoldIndex this is attributed to the set boundry condition (-b), pH and, most importantly, the pKa values used for the calculations.
- Coloured schema of disordered regions is properly rendered only in the terminal window and only if the terminal background colour is other than green or red.
- Aretas Gaspariunas
This project is licensed under the BSD License - see the LICENSE file for details
preFold is an original work and does not copy any code elements or principles of FoldIndex source code. It was built/cloned only using ideas presented in the FoldIndex (Prilsuky and Felder et al, 2005) and the foldability prediction algorithm (Uversky et al, 2000) publications, preFold was further improved using these ideas as a foundation. The peptide charge calculation method is used as described by Moore, 1985.