PrimerServer2: a high-throughput primer design and specificity-checking platform
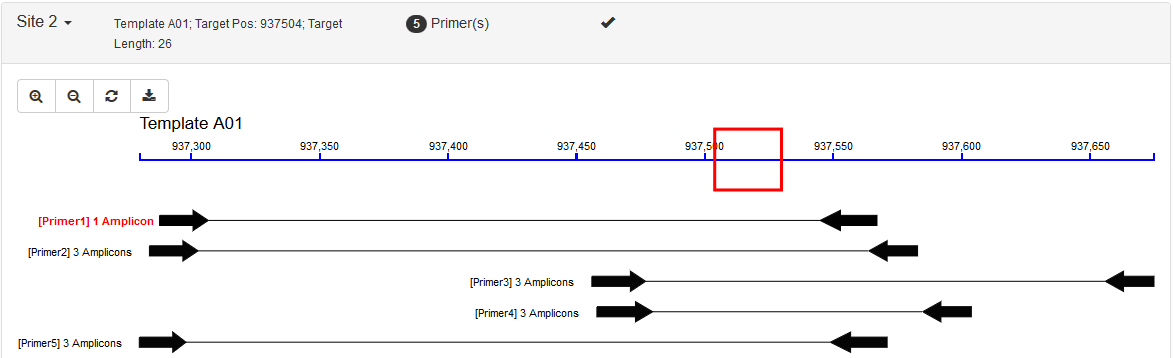
PrimerServer was proposed to design genome-wide specific PCR primers. It uses candidate primers produced by Primer3, uses BLAST and nucleotide thermodynamics to search for possible amplicons and filters out specific primers for each site. By using multiple threads, it runs very fast, ~0.4s per site in our case study for more than 10000 sites.
This repository is based on Python3 and acts as the successor of legacy PrimerServer.
Add these two softwares to your system PATH
- Samtools (>=1.9).
- NCBI BLAST+ (>=2.2.18)
Don't use Python 3.9 or above since the primer3-py module hasn't supported Python 3.9 yet.
conda create -n primer python=3.8
conda activate primer
$ pip3 install primerserver2
$ git clone https://github.com/billzt/PrimerServer2.git
$ cd PrimerServer2
$ python3 setup.py install
** (if installed from pip,) tests/query_design_multiple and tests/example.fa can be obtained from this github repository.
** full mode: design primers and check specificity
$ primertool full tests/query_design_multiple tests/example.fa
** design mode: design primers only
$ primertool design tests/query_design_multiple tests/example.fa
** check mode: check specificity only
$ primertool check tests/query_check_multiple tests/example.fa
If you have parts of template sequences, you can directly input in FASTA format:
>site1
TGTGATATTAAGTAAAGGAACATTAAACAATCTCGACACCAGATTGAATATCGATACAGA
TACCCCAACTGCCGCCAATTCAACCGACCCTTCACCACAAAAAAACTAATATTTATCAGC
CAATA[GTTACCTGTGTG]ATTAATAGATAAAGCTACAAAAGCAAGCTTGGTATGATAGT
TAATAATAAAAAAAGAAAAAACAAGTATCCAAATGGCCAACAAAGGCTGTATCAACAAGT
>site2
ACCAGATTGAATATCGATACAGATACCCCAACTGCCGCCAATTCAACCGACCCTTCACCA
CAAAAAAACTAATATTTATCA[GC]CAATAGTTACCTGTGTGATTAATAGATAAAGCTAC
AAGCAAGCTTGGTATGATAGTATTAATAATAAAAAAAGAAAAACAAGTATCCAAATGGCC
Note there is a pair of square brackets [] indicating target in each sequences. It means primers should be put around the target. This is the default mode.
If you have genomic coordinates for each site (e.g. SNPs), you can input coordinates like:
seq1 200 10
seq1 400 10
It means that two sites (one site per line) are needed to design primers. The first site is in seq1 and starts in position 200 and the region length is 10 (means seq1:200-209). The second site is in seq1 and starts in position 400 and the region length is 10 (means seq1:400-409).
For details, see the wiki.
Please refer to the wiki.
If you use reference genomes with many unplaced scaffolds, be caution since such scaffolds with great homology with main chromosomes might influence your results. If possible, delete (some or all of ) these unplaced scaffolds. For the human genome, we recommend the no_alt_analysis_set, which has all the PAR regions marked with N, to be used.
| CLI | Web UI | |
|---|---|---|
| Design primers | ✔️ | ✔️ |
| Checking specificity | ✔️ | ✔️ |
| Progress monitor | ✔️ | ✔️ |
| Number of tasks | High | Low |
| Alternative isoforms | ✔️ | ❌ |
| Exon-exon junction | ✔️ | ❌ |
| Pick internal oligos | ✔️ | ❌ |
| Custom Tm temperature | ✔️ | ❌ |
| Custom max amplicons | ✔️ | ❌ |
| Visualization | ❌ | ✔️ |